geneXplain platform news
Release 7.4
HOMER integration:
This release of the geneXplain platform introduces integration of the HOMER tools into the Galaxy section of the platform’s methods.
You will find two new motif discovery tools available under the Analyses → Galaxy → NGS: HOMER
1) The FindMotifs tool allowing you to analyze a Human, Mouse, or Rat input gene set in the Ensembl format
2) FindMotifsGenome tool allowing processing of BED files (in hg38, mm10, or rn6 genome builds of human, mouse, or rat).
Database updates:
- HumanPSD™ is updated to version 2024.1 (July 2024).
- TRANSFAC® is updated to version 2024.1 (July 2024).
- TRANSPATH® is updated to version 2024.1 (July 2024).
View full new features list here.
Release 7.3
Gene regulatory networks construction via API:
New Jupyter notebook Python sample code that allows construction of gene regulatory networks using the geneXplain platform API can be viewed from Colab notebook or downloaded from here.
Database updates:
- HumanPSD™ is updated to version 2023.2 (December 2023).
- TRANSFAC® is updated to version 2023.2 (December 2023).
- TRANSPATH® is updated to version 2023.2 (December 2023).
View full new features list here.
Release 7.2
Database updates:
- HumanPSD™ is updated to version 2023.1 (July 2023).
- TRANSFAC® is updated to version 2023.1 (July 2023).
- TRANSPATH® is updated to version 2023.1 (July 2023).
New workflows and methods:
- The platform now provides a new section called“Combinatorial regulation”, which provides tools for analysis of combinations of TF binding sites in promoters, enhancers and silencers, and other regulatory genomic regions. In this section, in addition to our traditional Composite Module Analyst, we introduce a completely new branch of site search combinatorial analysis methods based on the sparse logistic regression (MEALR).
- The new workflow Combinatorial regulation analysis of genomic or custom sequences scans input sequences for tissue and cell type- specific transcription factor binding regions using the newly created library of MEALR models delivered with the TRANSFAC® database.
- The new method MEALR combinatorial regulation analysis applies combinatorial regulatory models (CRMs) based on the MEALR affinity score to classify or scan sequences for occurrences of combinations of transcription factor binding sites represented by TRANSFAC® PWMs.
- The new method Extract TRANSFAC(R) PWMs from combinatorial regulation analysis extracts TRANSFAC® PWMs from a result table generated by the MEALR combinatorial regulation analysis. The PWMs represent transcription factor binding motifs for the TF in focus and its co-factors that constitute the combinatorial module predicted by the MEALR model.
New import possibility:
- The files from the Sequence Read Archive (SRA) can be now uploaded to the geneXplain platform directly by the SRR ID.
Release 7.1
Database updates:
- HumanPSD™ is updated to version 2022.2 (November 2022).
- TRANSFAC® is updated to version 2022.2 (November 2022).
- TRANSPATH® is updated to version 2022.2 (November 2022).
Enhancements:
- The platform now supports the following species for performing a vast number of analyses including search for TFBS on an input gene set: FruitFly, Nematoda, Saccharomyces Cerevisiae (baker’s yeast), Schizosaccharomyces Pombe (fission yeast).
- All geneXplain platform related videos with analysis demo and how-to instructions were now accumulated in a new Videos page
Platform APIs (genexplain-api and geneXplainR) now have extended documentation and tutorial freely available online at this page. Additional information on geneXplain platform API, as well as the how-to videos on its usage with demo scripts, can be now found on this page.
Release 7.0
New:
- The platform has now fully automatic pipelines for paired RNA-seq libraries
- New workflows for analyzing multiple BAM files
- Estimate DEGs from raw RNA-seq data, counts or normalized transcriptomic data in 8 new and enhanced workflows
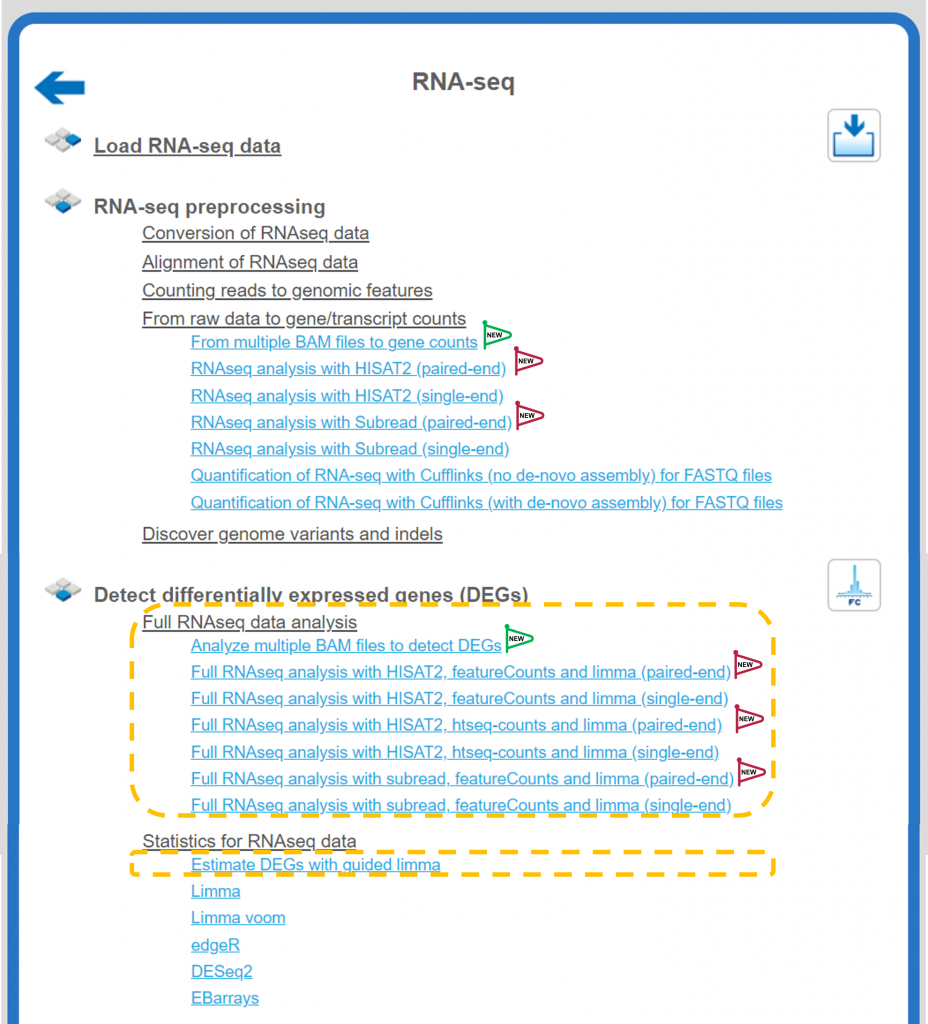
- Support of new species Zebrafish and Arabidopsis for analyzing RNA-seq data or genes and sequences for transcription factor binding sites in over 20 workflows

-
- Support of Affymetrix miRNA-4.1 microarray chip & Agilent miRNA microarray chips
- Enable multiple miRNA ID support and conversion handling
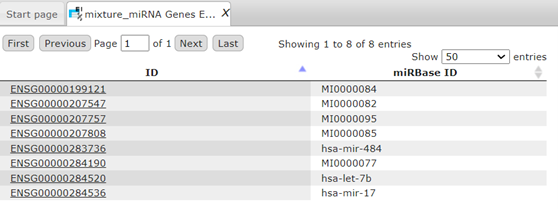
- Investigate tissue- or cell-line specific miRNA promoter regions with TRANSFAC®
Identify enriched motifs in tissue-specific miRNA promoters [ workflow link ]
Identify enriched motifs in cell-specific miRNA promoters [ workflow link ]
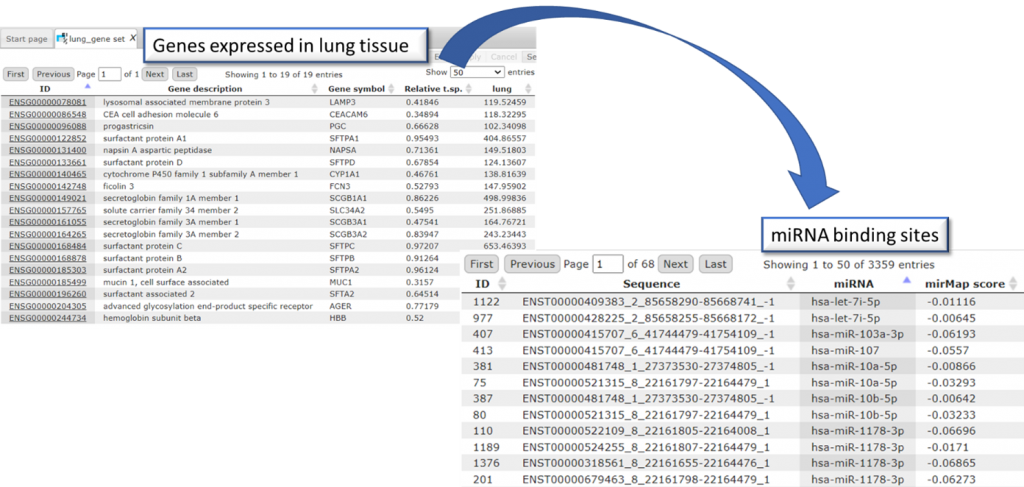
- Identifying miRNA binding sites in tissue-specific genes with HumanPSD™
- New pipeline to analyze SNPs with TRANSFAC® and TRANSPATH® at once

- New features: MATCH™ for tracks, Convert table to VCF track, Filter VCF by genotype
- We have updated our start page with new features and > 30 enhanced or new workflows

Database updates:
- HumanPSD™ is updated to version 2022.1
- TRANSFAC® is updated to version 2022.1
- TRANSPATH® is updated to version 2022.1
Release 6.5
Database updates:
- HumanPSD™ is updated to version 2021.3 (December 2021).
- TRANSFAC® is updated to version 2021.3 (December 2021).
- TRANSPATH® is updated to version 2021.3 (December 2021).
Enhancements:
- The platform supports now the new Ensembl Mus musculus genome assembly GRCm39 (mm39). An example can be seen here.
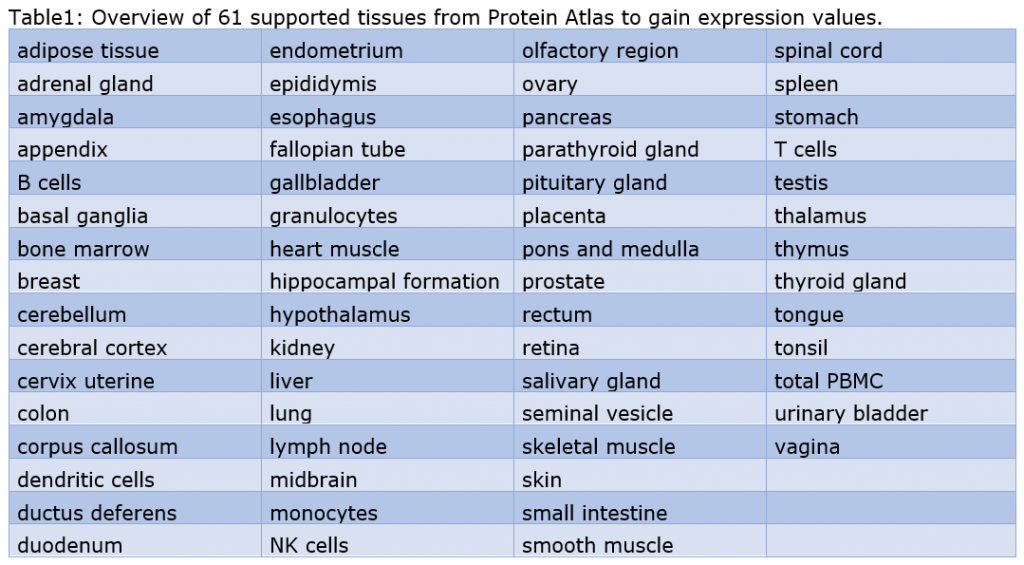
- Human Protein Atlas data is now incorporated into the platform to allow queries for tissue specific expression values of genes. We are supporting 61 different tissues. The output gene lists can be further filtered for 14 different specified protein functions and contain the corresponding annotation information.

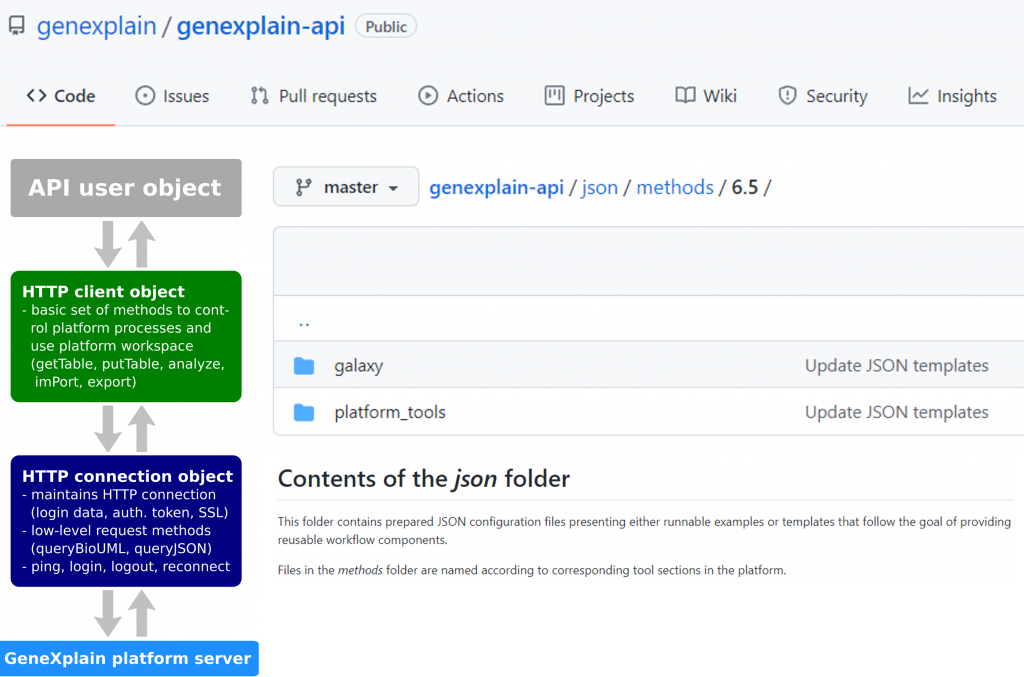
- Platform APIs (genexplain-api and geneXplainR) were updated to provide all method parameters including expert options. JSON templates cover all platform as well as the integrated Galaxy tools of edition 6.5.
Release 6.4
Database updates:
- HumanPSD™ is updated to version 2021.2 (September 2021).
- TRANSFAC® is updated to version 2021.2 (September 2021).
- TRANSPATH® is updated to version 2021.2 (September 2021).
Enhancements:
- Transfac ‘Matrix to Ensembl’ matching was extended by links from matrices to orthologous and paralogous Ensembl identifiers. These links are used in hub, which converts Matrices to Ensembl genes in Convert table analysis.
- New proteomics example with Gene Expression Omnibus data (GSE66789): Study of Myc induced regulation of protein translation. Find it at Data –> Examples –> Proteome profiling upon Myc activation, GSE66789.
- Extended descriptions were added for the following examples:
· COVID-19 suppress innate immune responses GSE156063, Illumina high throughput sequencing
· Case study for RNA-seq data analysis
· Chronic Myeloid Leukemia Patient Genotyping
· Cytokine-triggered gene expression in cell cycle stages, GSE52465, Agilent-014850 microarray
· E2F1 binding regions in HeLa cells, ChIP-Seq
· HCV infection in liver GSE31193, Affymetrix U133 Plus 2.0 microarray
· Proteome profiling upon Myc activation, GSE66789
· TNF-stimulation of HUVECs GSE2639, Affymetrix HG-U133A microarray
· miRNA regulation by Myocardin and ERalpha, GSE44139, Affymetrix Multispecies miRNA-2
Release 6.3
Database updates:
- HumanPSD™ is updated to version 2021.1 (January 2021).
- TRANSFAC® is updated to version 2021.1 (January 2021).
- TRANSPATH® is updated to version 2021.1 (January 2021).
New workflow:
- Identify enriched motifs in cell specific promoters (TRANSFAC(R))
This workflow searches for enriched transcription factor binding sites (TFBSs) in a set of gene promoters versus a random promoter set. The input gene set is used to extract promoter regions by mapping it against the TSS locations defined in the Fantom5 (Nature 507:462–470) database for one selected cell-type among 172 available cell-types. The over-represented sites identified with the MEALR method are converted into a profile, which is used for a second round of site search analysis and ends up with the identification of potential transcription factors.
New features:
- MEALR classifier (tracks)
MEALR searches for a combination of transcription factor binding motifs that discriminate between a positive (Yes) and a negative (No) sequence set. This tool takes a sparse logistic regression model derived with MEALR and applies it to new sequences to predict whether they can be bound by TF complexes or contribute to gene regulation in the same way as the Yes sequences used to train the MEALR model.
- Random forest prediction
Random forests are a combination of tree predictors and that each tree depends on the values of a random vector sampled independently and with the same distribution for all trees in the forest. This statistical method performs a classification or regression with a random forest model, based on Breiman (published in Machine Learning). Please refer to documentation of the R randomForest package for computational details. Random forests can be trained using the Train random forest tool (see below).
- Train random forest
This method can be used to train a random forest model for classification, regression, or clustering. Please refer to documentation of the R randomForest package for computational details. Except for unsupervised models, the trained random forests can be used for further classification or regression analysis with the Random forest prediction tool.
- t-SNE
The new tool contains a R wrapper around the fast T-distributed Stochastic Neighbor Embedding implementation by Van der Maaten (more information on the original implementation is here). The tool can be used for data visualization using the t-SNE algorithm.
Enhancements:
- Fantom5 workflows now available for both, hg19 and hg38 genome versions.
- The workflow for analyzing a SNP list with TRANSFAC database is now available for the hg38 genome version.
- New example with Gene Expression Omnibus data (GSE156063): Upper airway gene expression differentiates COVID-19 from other acute respiratory illnesses and reveals suppression of innate immune responses by SARS-CoV-2, Expression profiling by high throughput sequencing, Illumina NovaSeq 6000 Homo sapiens.
Release 6.2
Databases update:
- TRANSFAC®, TRANSPATH® and HumanPSD™ databases update to release 2020.3
- Ensembl version update to release 100
- Reactome database is updated to version 74
New workflow:
Compute differentially expressed genes using Limma and Metadata
This workflow performs a linear model analysis to identify differentially expressed genes from multiple samples using Limma statistics and a metadata table for the samples. The given input table contains expression values from several samples and a corresponding sample table (metadata) for guiding the limma analysis by selected experimental factors. The workflow aims at finding significant differences between pairs of levels of a main factor (Treatment). Furthermore, an ANOVA is carried out for all contrasts together. The primary result of the linear model analysis is further filtered to identify significant up- and down-regulated genes for each sample comparison.
New features:
Join full tables
Joining two tables into a new one with containing the selected columns. Different joining types can be processed according to the ID matching from both input tables. Following joining types are available: Inner join, outer join, left join, right join, left subtraction, right subtraction, and symmetric difference.
Calculate keynodes ranks
This method adds new score-specific ranks for each identified master regulator molecule to identify the best corresponding master regulator from the input list.
Select keynodes with top targets
This method selects the top master regulators (keynodes) by score and linkage to top targets, which can be potential drugs from HumanPSD database.
Enhancements:
- Update MTB report to version 2.0.0
- Removal of redundant Ensembl versions for the same build
- Add Ensembl annotation source to all workflows
- Bug fixing of Affymetrix miRNA chips normalization
- Easy selection of current data project
RELEASE 6.0
Databases update:
- TRANSFAC®, TRANSPATH® and HumanPSD™ databases update to release 2020.2
- Ensembl version update to release 99
- Gene Ontology update to version 2020-03-25
New features:
- New Import – import now supports the Drag and Drop function.
- EdgeR for two tables – method now supports two separate read counts tables for samples and control samples.
- MPT report – search within curated databases (GKDB1 and CIViC2) for predictive biomarkers according to their clinical evidence for somatic variants (mutations, amplifications, deletions, rearrangements) of a patient and outputs automatic pdf report.
- Calculate CMA regulation – calculates regulatory scores for transcription factors from a CMA (composite modules analysis) and Master regulator (MR) analysis result and creates a visualization of top MRs with CMA modules, underlying genes and potential feed forward loop regulation.
- Create profile from CMA model – generates a matrix collection of transcription factors found in a user’s CMA model.
- Find regulatory regions with mutations – calculates mutation scores using information about mutation locations.
- PSD pharmaceutical compounds analysis – generates a table with drugs known to be acting against the corresponding targets of an input gene list.
New workflows:
Workflows allow a fully automatic analysis from raw RNAseq reads with pre-processing, alignment summaries, quality control, plots and estimation of differentially expressed genes (DEGs) for unlimited FASTQ files stored simply in a data folder:
- Full RNAseq analysis with HISAT2, featureCounts and limma
- Full RNAseq analysis with HISAT2, htseq-counts and limma
- Full RNAseq analysis with subread, featureCounts and limma
Enhancements:
- Transparent genome version control – easy selection of a genome version in RNAseq pre-processing pipelines, which will be applied to state-of-the-art alignments methods.
For more details please explore the full new features list of geneXplain® platform release 6.0.
RELEASE 4.9
- New start page icons in any of the platform’s research categories
- Pre-release of TRANSFAC®, TRANSPATH® and HumanPSD databases (version 2018.2)
- Update to Ensembl 91 database
- Update to Reactome 63 database
RELEASE 4.8
The geneXplain platform toolbox for bioinformatics data analysis contains these new functional features in the current release:
- New analysis methods
Construct composite modules on track (correlation) – method predicts composite module using the result of the “Site search on gene set” analysis.
Cluster track – method clusters sites in a track, what is useful for merging of closely spaced sites into one big cluster.
Compute profile thresholds – method computes profile thresholds minimizing either false negatives(minFN) or false positive(minFP) on the random DNA sequence.
Create miRNA promoters – method extracts miRNA promoters from mirprom database for a given list of miRNAs
Get transcripts track – method extracts track from a database by a transcript ID
Recalculate composite module score on new track – method takes best composite model from the given CMA result and calculates its scores on all sites of a given track.
Continue CMA – method continues prediction of composite module using results of the previous prediction as a start point. Prediction parameters are customizable.
Table Imputation – method replaces missing data in the given input table with row means.
- New HTML report for site search analysis
You can now create a summary of your site search analysis including visualization of input promoters together with identified enriched transcription factor binding sites (TFBSs) in HTML format, which can be exported to your local computer. These results can be easily used for presentations or publications.
- New toolbar buttons
Check out our new toolbar icons which will lead you to remarkable results in your research simply by a couple of clicks.
- Integration with updated TRANSFAC®, TRANSPATH® and HumanPSDTM databases in release 2018.1
The TRANSFAC® database of transcription factors, their genomic binding sites and DNA-binding motifs (PWMs), TRANSPATH® database of mammalian signal transduction and metabolic pathways and Human Proteome Survey Database (HumanPSDTM) with focus on human proteins as disease biomarkers and drug targets in their 2018.1 release versions are currently integrated with the geneXplain platform.
RELEASE 4.7
Installation of TRANSFAC 2017.3 (information download)
– Annotation of transcription factor binding sites based on sequence conservation
– ChIP-Seq experiment browse pages
– Reorganization of the in vivo transcription factor bound fragment section on a Locus Report
– HOCOMOCO v10 matrix library integration
– Enhanced human SNP content
– Ensembl version update
Installation of TRANSPATH & HumanPSD 2017.3 (information download)
– Integration of new clinical trial (CT) data sources
– Improved user data management
– Quick search for disease and drug entries
– Link-out to BRENDA professional – the comprehensive enzyme information system
– New phosphorylation targets content
RELEASE 4.6
New method: LRPath is a Gene Set Enrichment Analysis (GSEA) method that uses logistic regression models to discover categories that are significantly correlated with a predictor.
New protein category (TRANSPATH® isogroups) to enhance identification of master regulators.
RELEASE 4.5
Installation of TRANSFAC public:
– Available for everyone
– 219 profiles (matrices) for site search tools
– Search function implemented
DESeq tool:
– Bug fixed that prevented analysis from completing correctly
– Added option to run DESeq or DESeq2
– New versions of PROTEOMETM data now named HumanPSDTM database
– Latest release 2017.2 available in the geneXpain platform
– Platform Java API available from github.com/genexplain/genexplain-api
– Executable jar can be configured with JSON config files to invoke platform processes from the command line