Approaches to pathway analysis
To find out whether among all genes induced in an experiment those are overrepresented that encode components of a certain pathway, conventional gene set enrichment analysis (GSEA) and related methods can be applied. In such an approach, however, topological information about the pathway is lost.
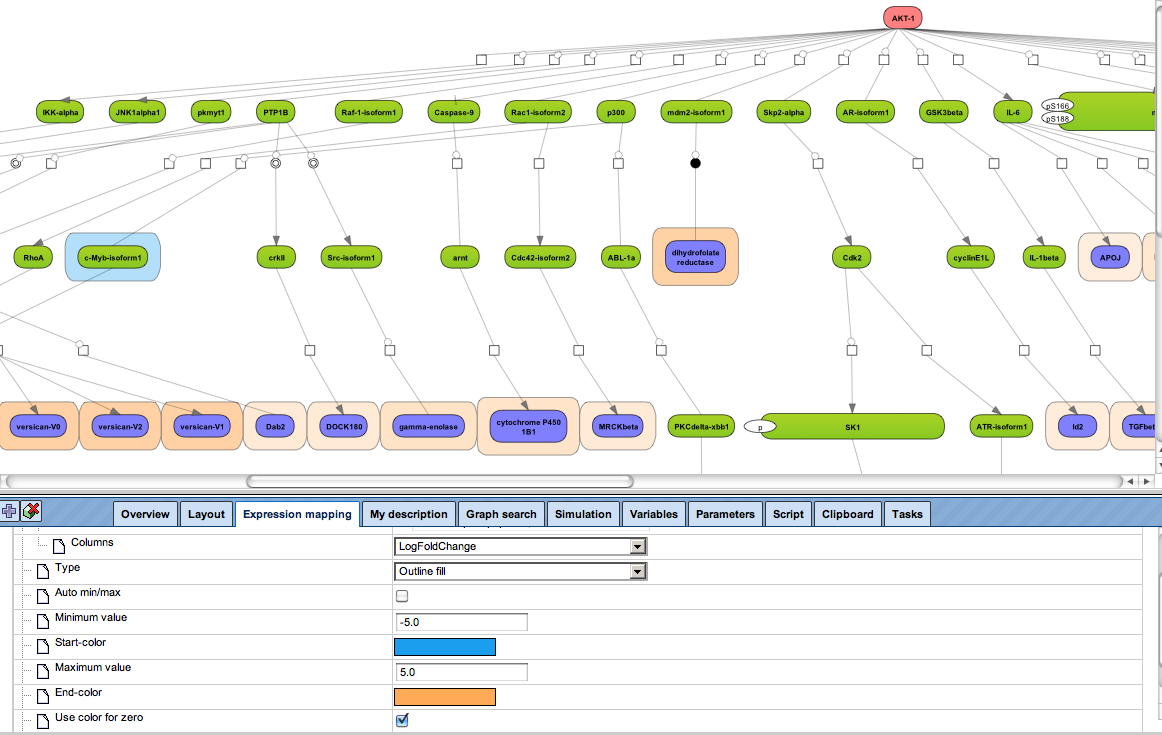
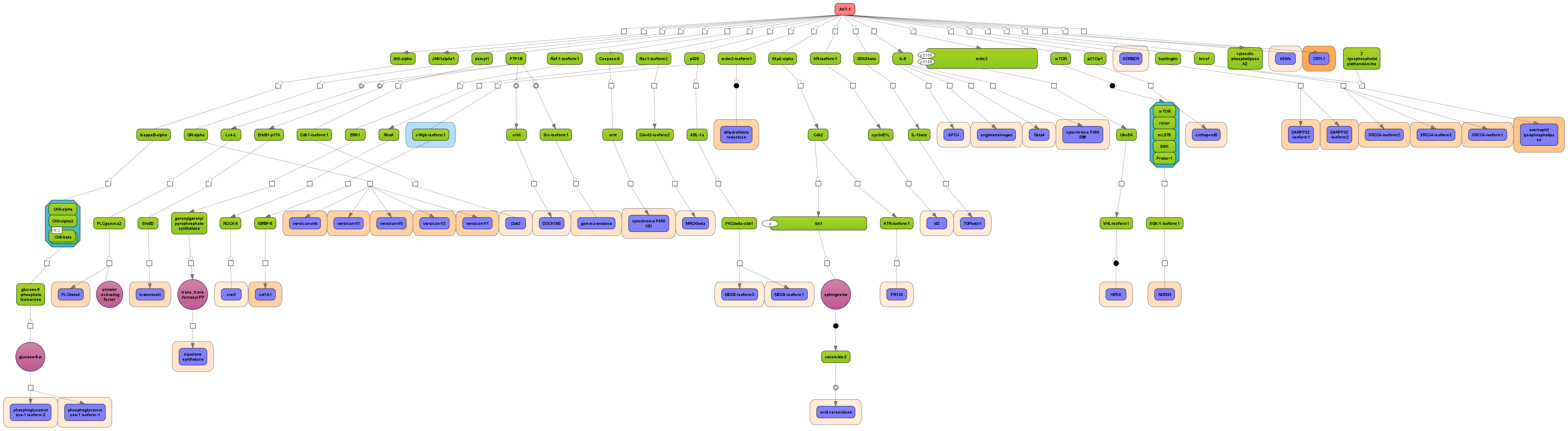
More sophisticated is to search for those networks, pathways or paths where many linked components have been induced. This is provided by the platform option “Cluster by shortest path”. A visualization of differential expression onto a known pathway is shown in the figure below. These known pathways may be documented in the databases TRANSPATH® (manually curated information; example shown) or GeneWays (compiled by text mining).
Learn more about the geneXplain platform.
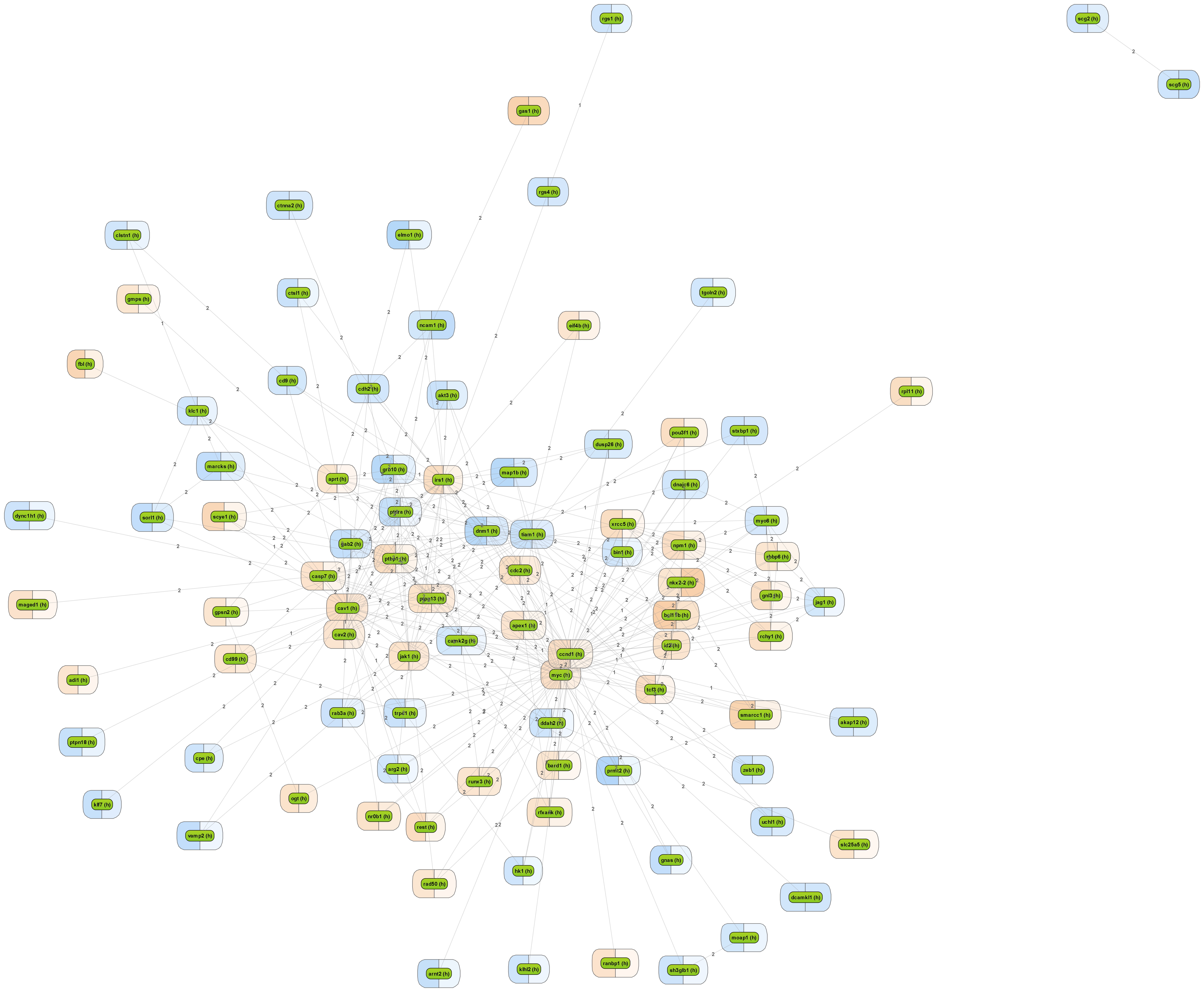
Click image for an enlarged view of the visualization of the clustered paths comprising a maximal number of differentially expressed genes, highlighted by a customizable color code between most significantly down- (blue) or up-regulated (orange) genes in two different experiments (left/right half of the colored boxes around the gene names). Edge labels indicate the number of steps between the shown nodes. In this example, mapping was done onto the GeneWays database network.
When starting from a set of differentially expressed genes or their products, resp., it is frequently of interest to see what is their common activator. Such convergence points of upstream pathways are potential master regulators, or key nodes.
The next figure shows how the upstream paths of a set of proteins (blue) converge in one master regulator (here: AKT1, red). The database behind this analysis is TRANSPATH®. It can be seen how a section of the whole pathway (overview in the lower right window) is amenable to editing in the main work area. Detailed information about a selected component, like the mTor complex in this example, are displayed in the Info Box at the lower left corner.
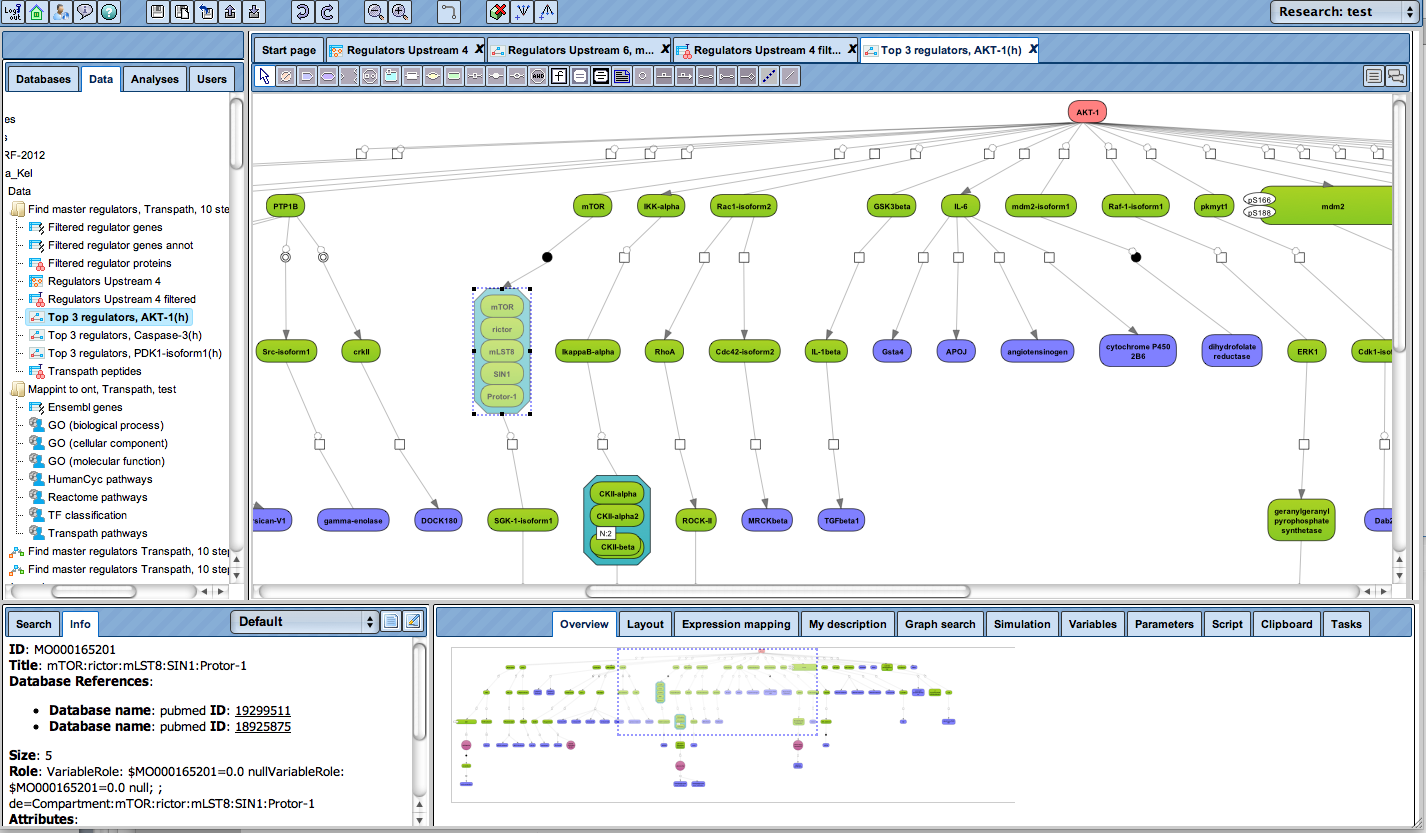
This type of analysis can be combined with the visualization of differentially expressed genes and their expression behavior, in the same way as shown above.