Stegmaier P., Voss N., Meier T., Kel A., Wingender E., Borlak J. (2011) Advanced computational biology methods identify molecular switches for malignancy in an EGF mouse model of liver cancer. PLoS ONE 6, e17738. PubMed.
Stegmaier P., Krull M., Voss N., Kel A., Wingender E. (2010) Molecular mechanistic associations of human diseases. BMC Syst. Biol. 4, 1024. PubMed.
Zubarev R.A., Nielsen M.L., Fung E.M., Savitski M.M., Kel-Margoulis O., Wingender E., Kel A. (2008) Identification of dominant signaling pathways from proteomics expression data. J. Proteom. 71, 89-96. PubMed.
Michael H., Hogan J., Kel A., Kel-Margoulis O., Schacherer F., Voss N., Wingender E. (2008) Building a knowledge base for systems pathology. Brief. Bioinform. 9, 518-531. PubMed.
Kel A., Voss N., Jauregui R., Kel-Margoulis O., Wingender E. (2006) Beyond microarrays: Find key transcription factors controlling signal transduction pathways. BMC Bioinformatics 7 Suppl. 2, S13. PubMed.
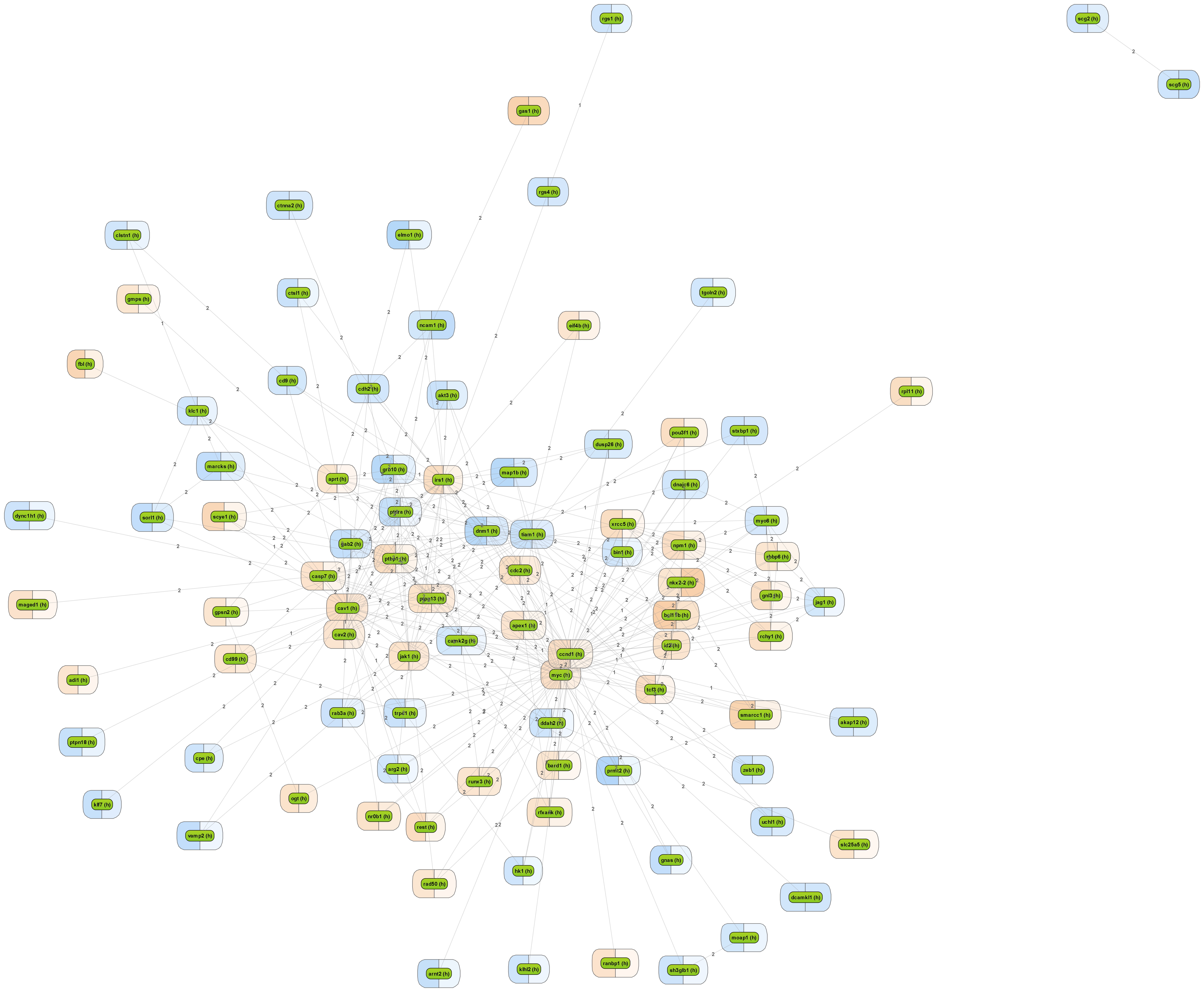
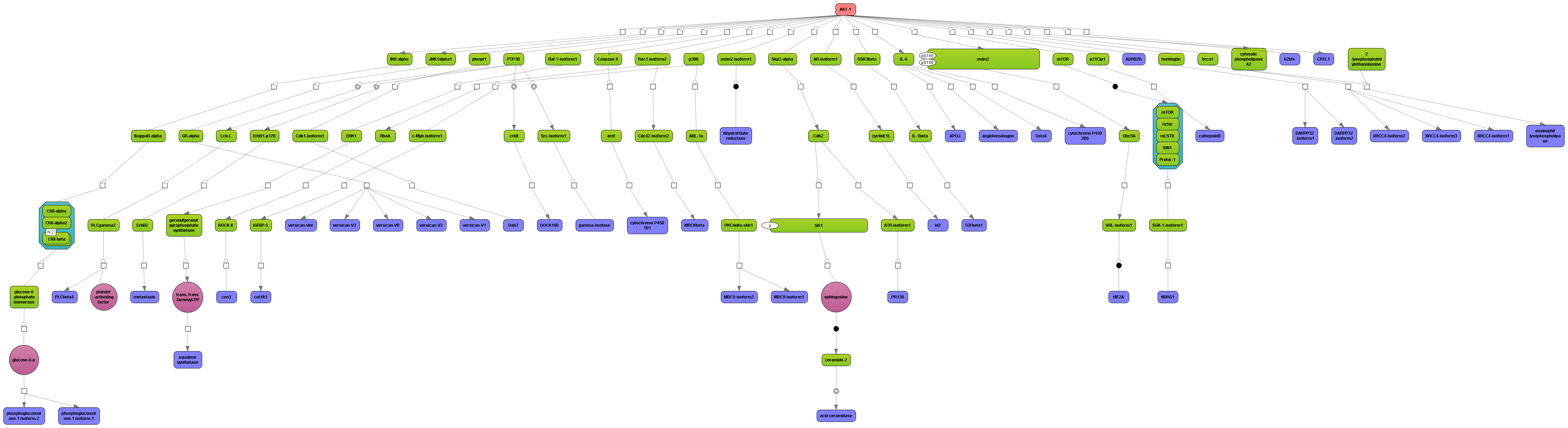
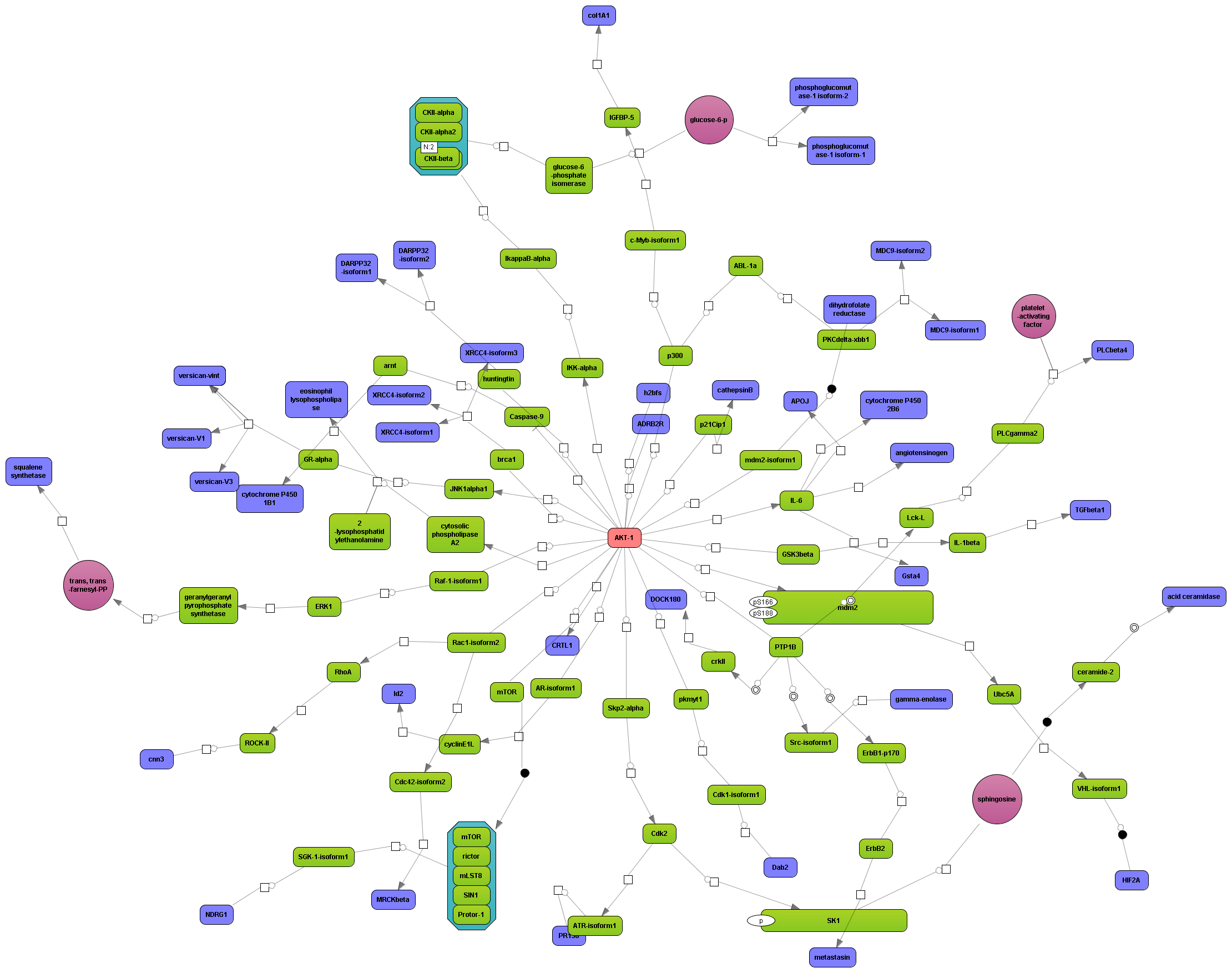
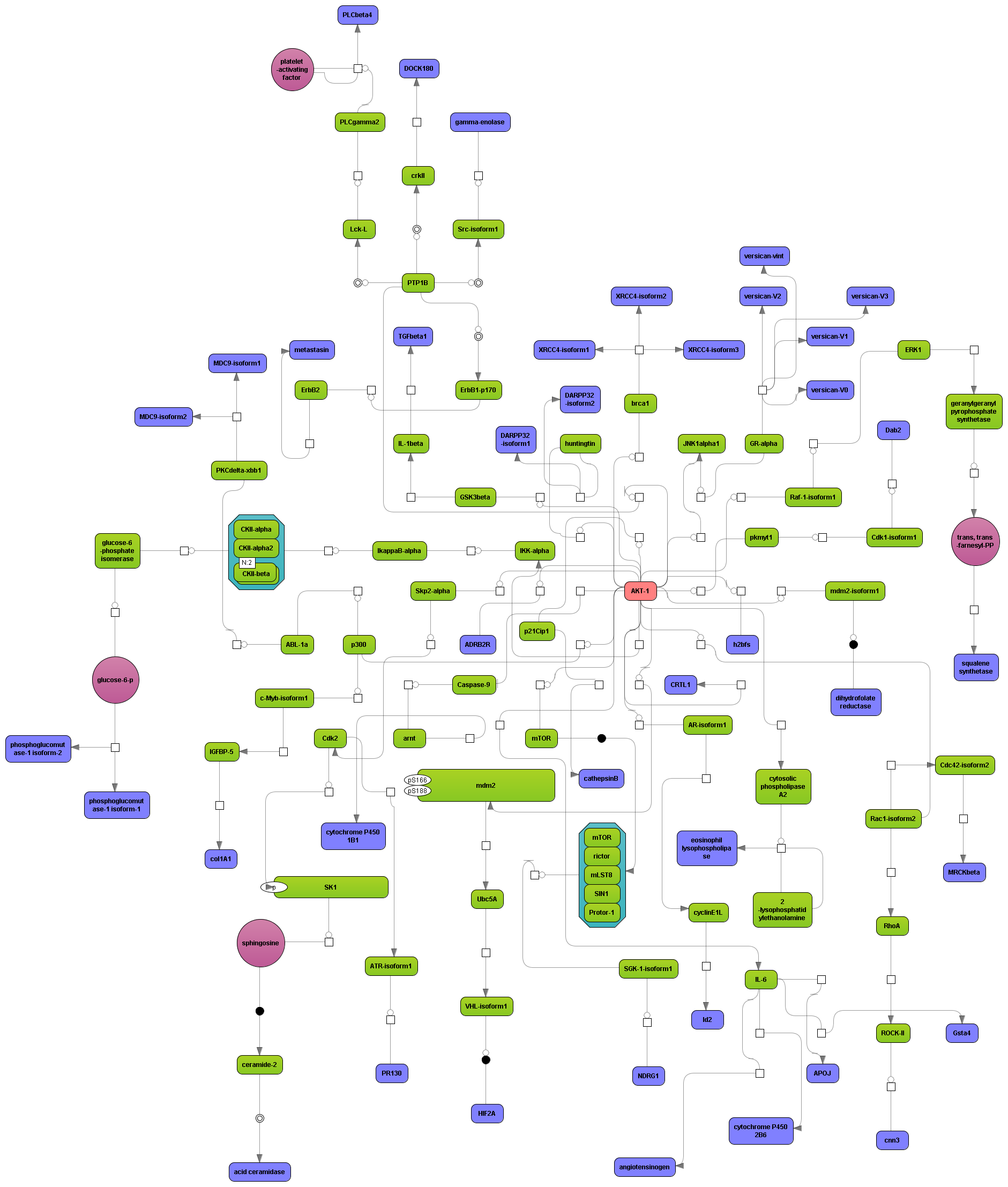
Get a fast overview about the pathways affected in your experiment.
Get an immediate indication about common effector molecules.
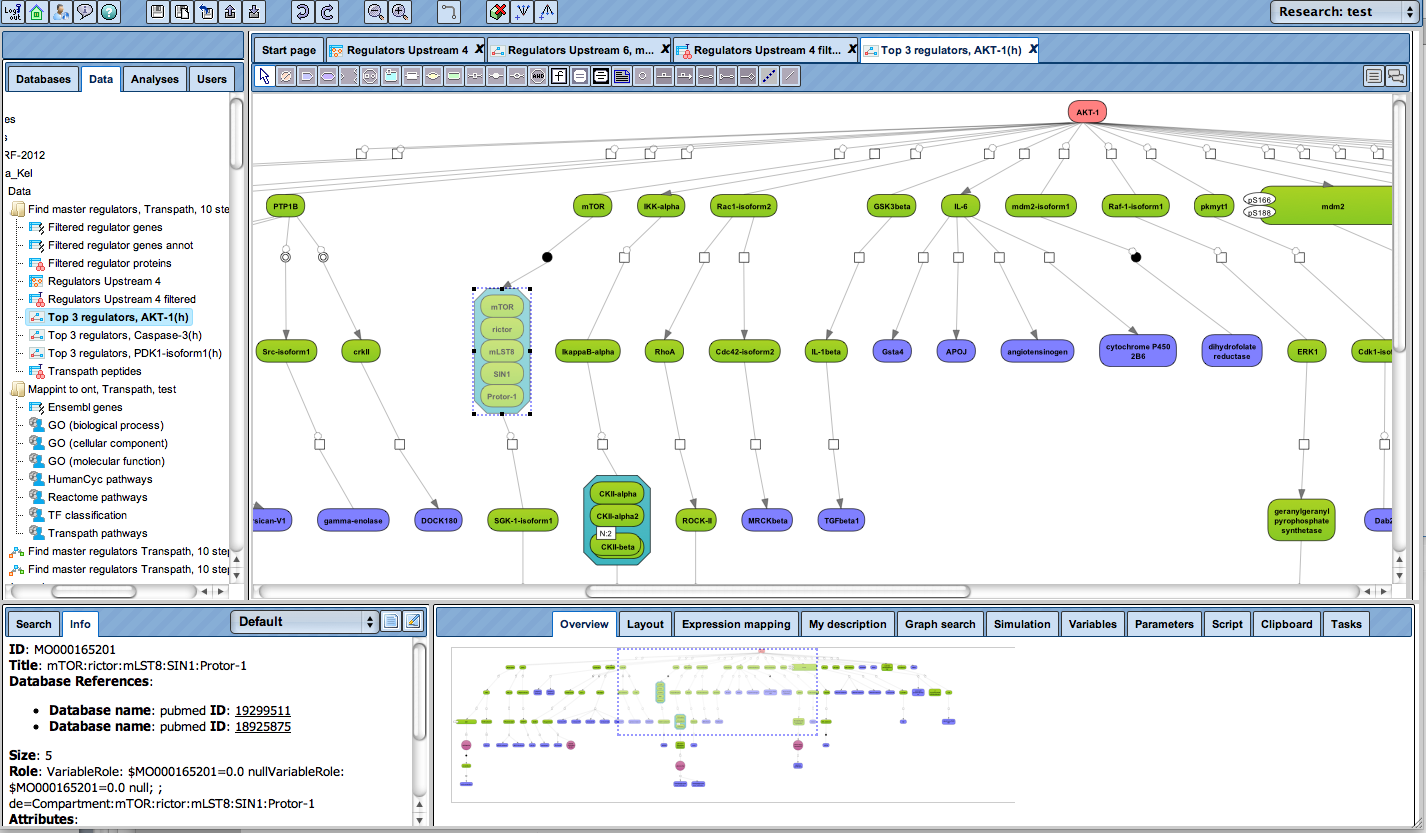
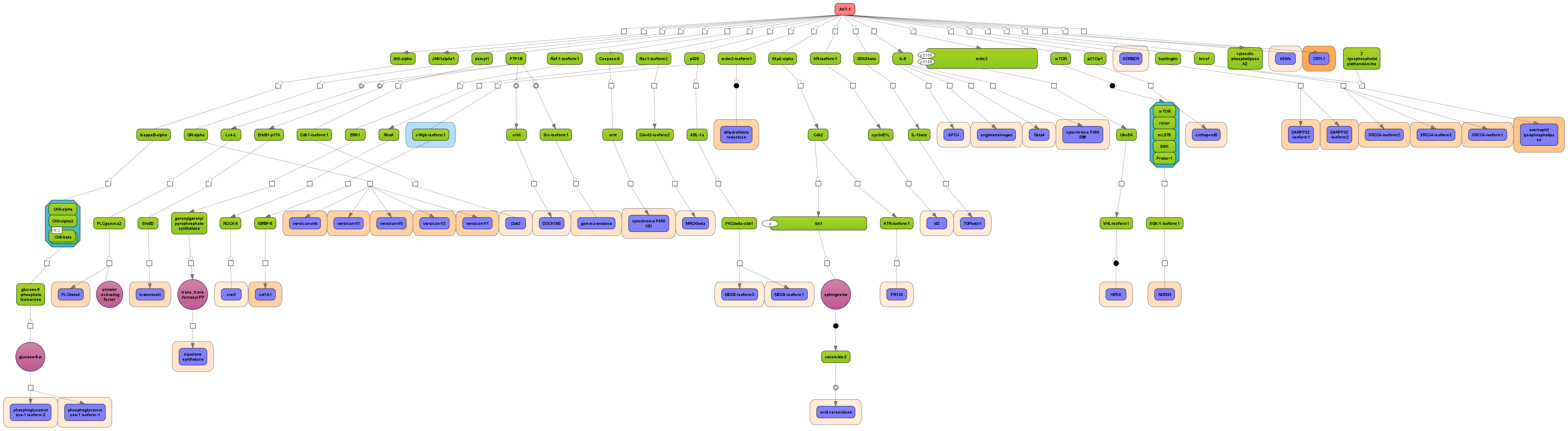
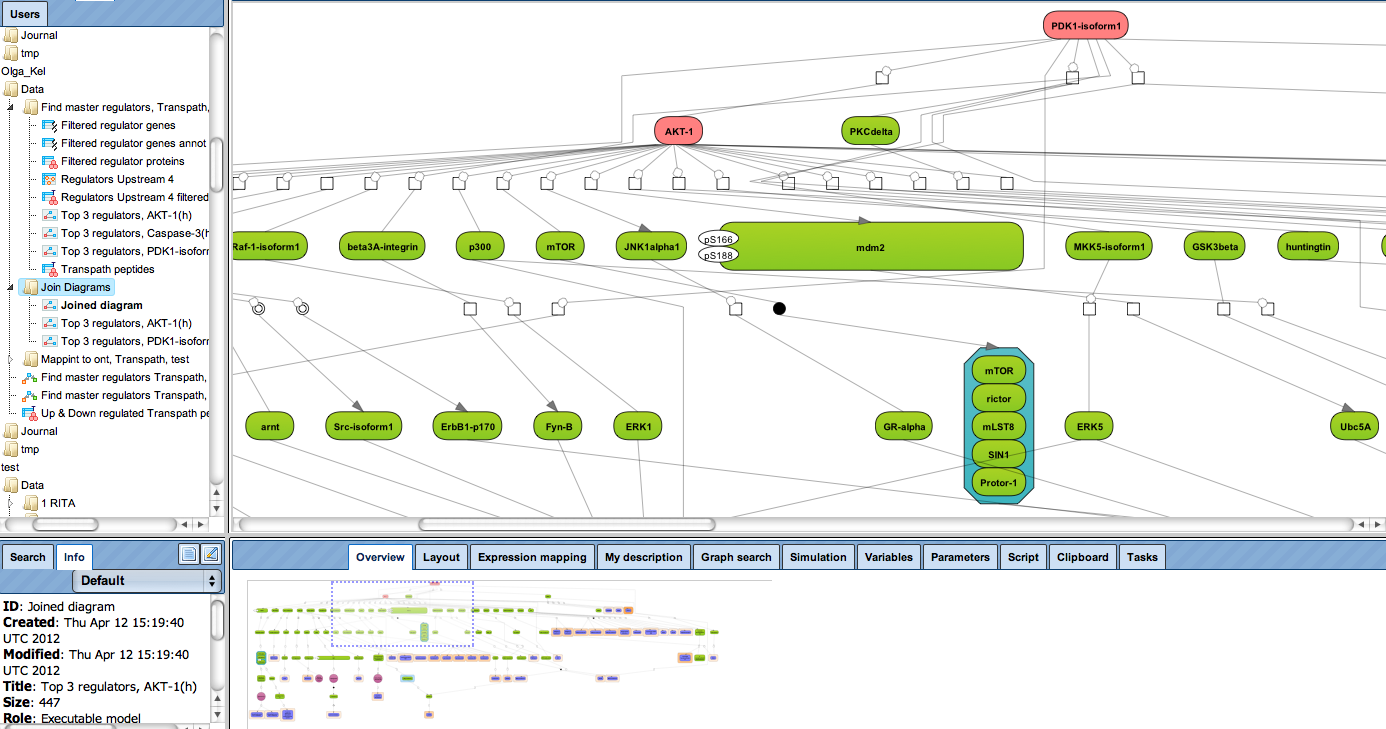
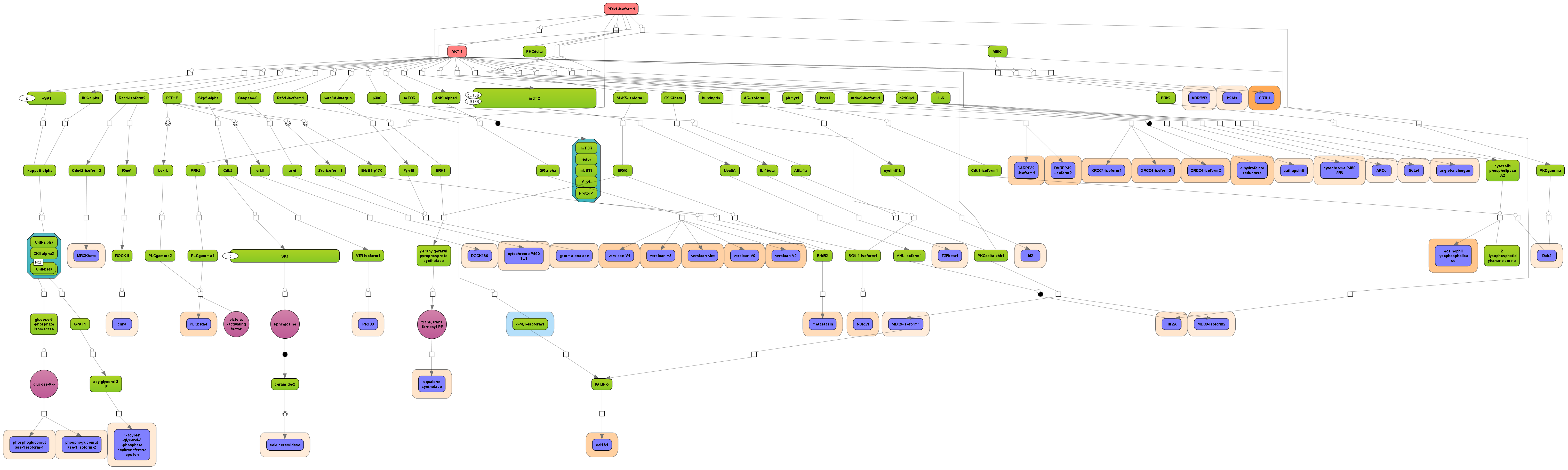
Receive a reliable prognosis about potential upstream master regulators.
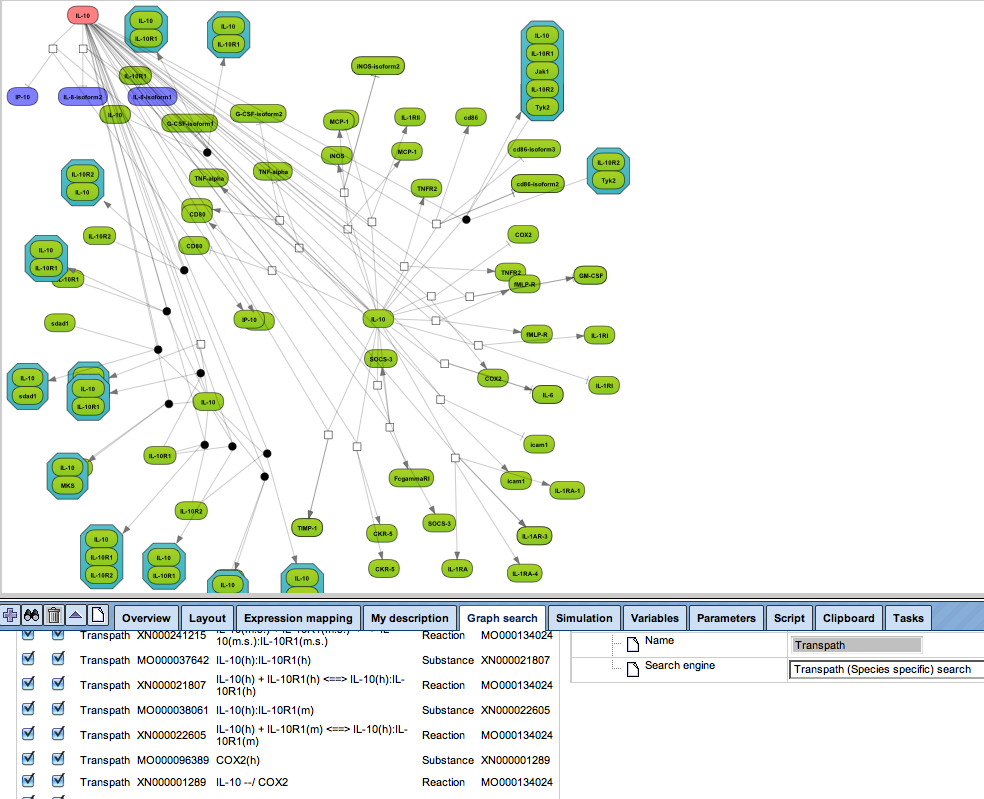
Flexibly work with manually curated pathway data (TRANSPATH) or with information automatically extracted from scientific literature (GeneWays).
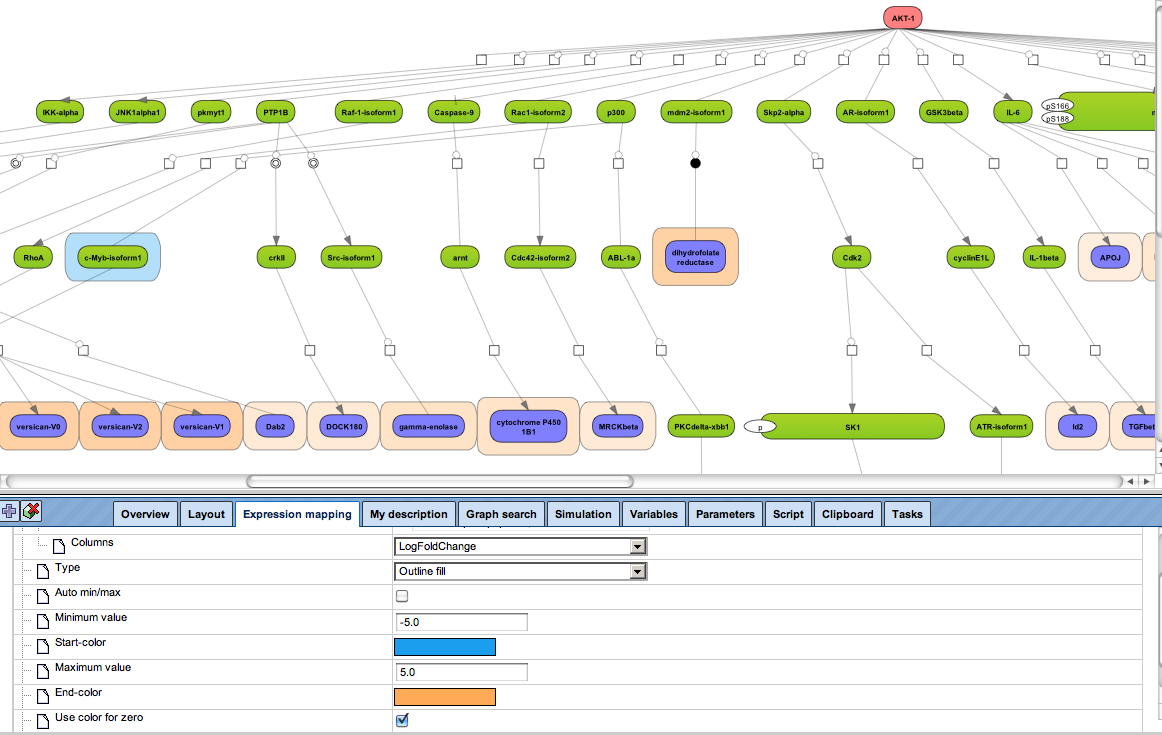
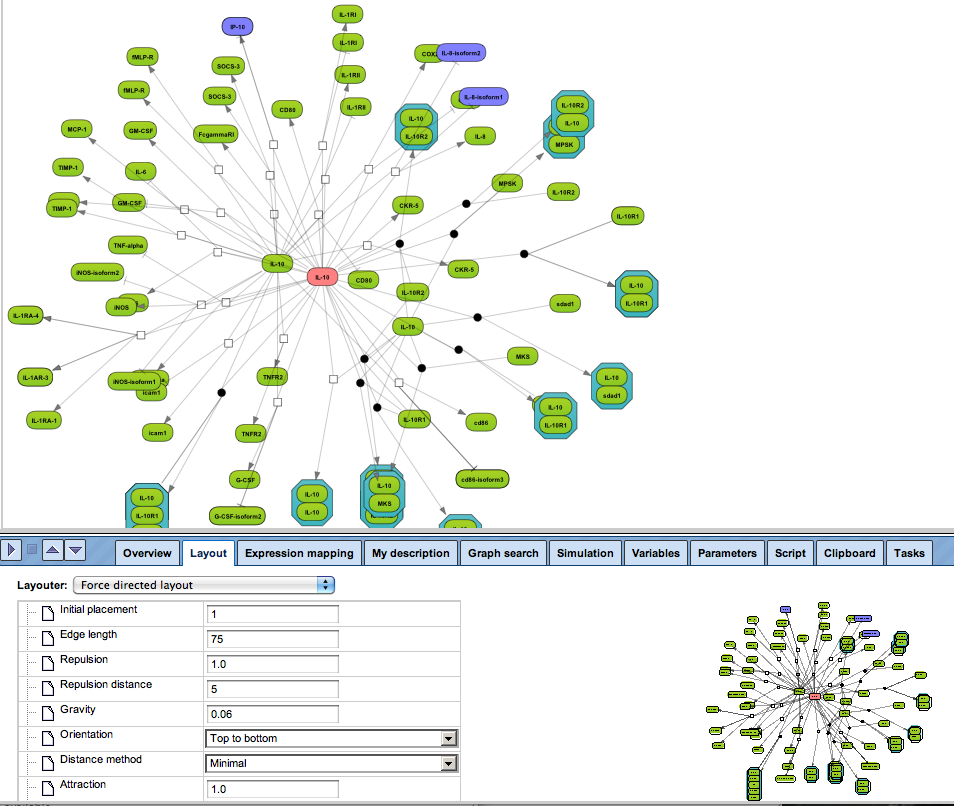
Enjoy variable layout, expansion and joining functions.
Visualize your network according to internationally accepted SBGN standards (Systems Biology Graphical Notation).
Benefit from high-quality and flexible export routines for your publications.