Upstream analysis:
The TRANSFAC PATHWAYS package uniquely combines promoter analysis with pathway analysis, enabling the identification of master regulators in gene regulatory networks. No other tool on the market provides such an integrated capability.
What is Upstream Analysis?
GeneXplain’s proprietary approach to analyze gene expression data is called Upstream Analysis. The term indicates that it is a causal analysis, providing a clue about the reason why a certain set of genes has been up- (or down-) regulated in the system under study. In contrast, conventional analyses usually reveal the effects of the differentially expressed genes, e.g. by mapping them onto ontological categories.
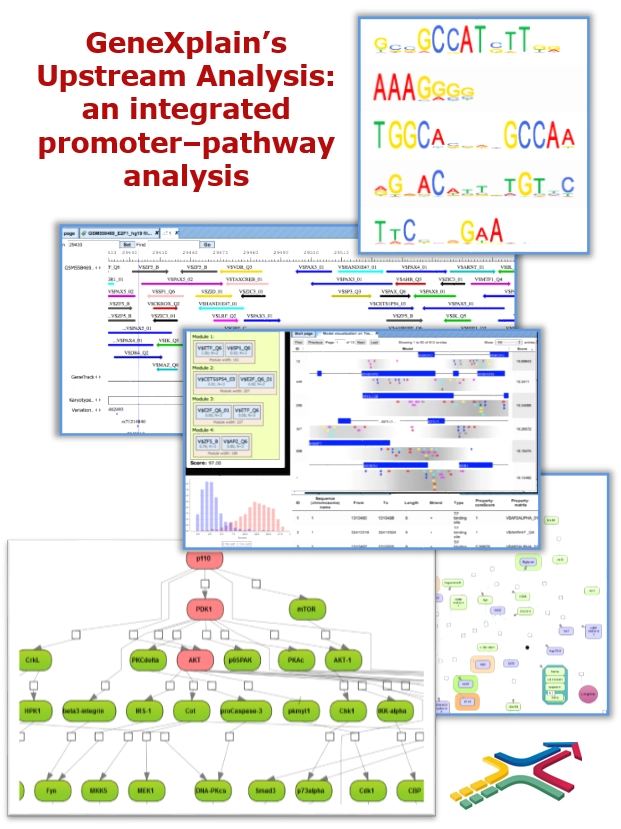
How does it work?
GeneXplain’s Upstream Analysis is an integrated promoter – pathway analysis. It starts from any list of differentially expressed genes (DEGs), which you may have extracted from your raw data with the aid of the geneXplain platform, and comprises two main steps:
- At first, the promoters of the differentially regulated genes are retrieved and analyzed for potential transcription factor (TF) binding sites and their combinations. From that, a set of TFs is identified that potentially have regulated the found DEGs.
- In a second step, the pathways are reconstructed that are known to activate the previously hypothesized TFs. Molecules where these pathways converge are considered as potential master regulators of the process under study
Step 1: Promoter analysis
First, potential transcription factor binding sites (TFBSs) are identified in all promoters of the DEGs of your experiment (Yes set) as well as in a negative control set (No set). This is the usually done with a library of position-specific scoring or positional weight matrices (PSSMs or PWMs).
We recommend to apply the most comprehensive matrix library available, the TRANSFAC® database, and using the MATCHTM algorithm for the sequence analysis.
Next, out of all these potential transcription factor binding sites (TFBSs), those that are characteristic for the DEG set under study are identified. This is done by rigorously determining their enrichment in the Yes- compared to the No set.
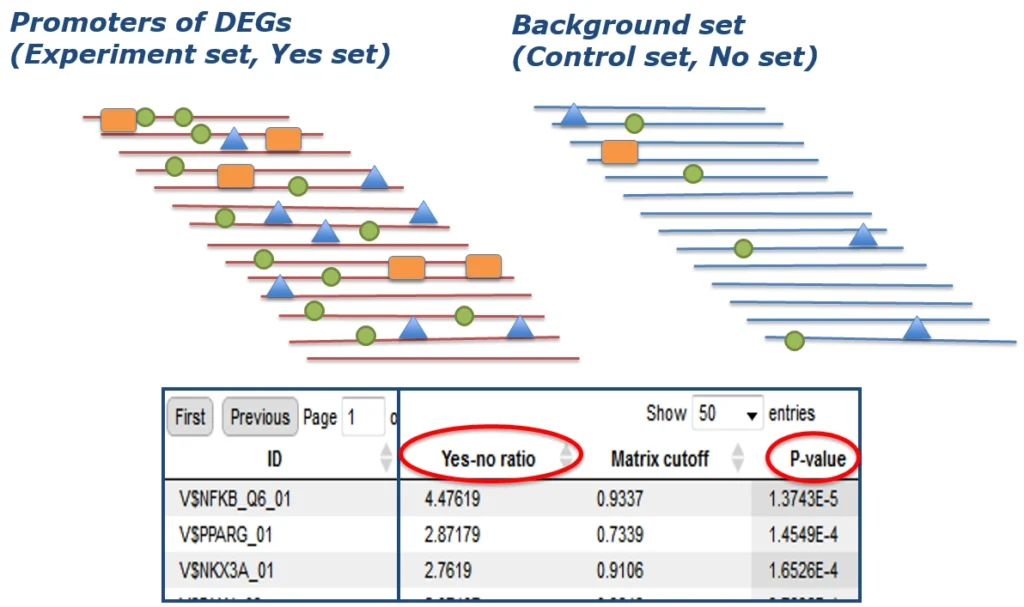
Learn more about promoter analysis with TRANSFAC® in the geneXplain platform.
Step 2: Pathway analysis
Step 1 resulted in a set of transcription factors (TFs), that are likely repsonsible for the differential regulation of the observed set of DEGs. From available pathway data, we have extracted information about all relevant signaling cascades that regulate the activity of TFs; optimally, the TRANSPATH® database is used for this and the further analysis.
As has been proven in a large number of use cases, these pathways usually converge in a couple of key nodes, which qualify as candidate master regulators of the process under study.
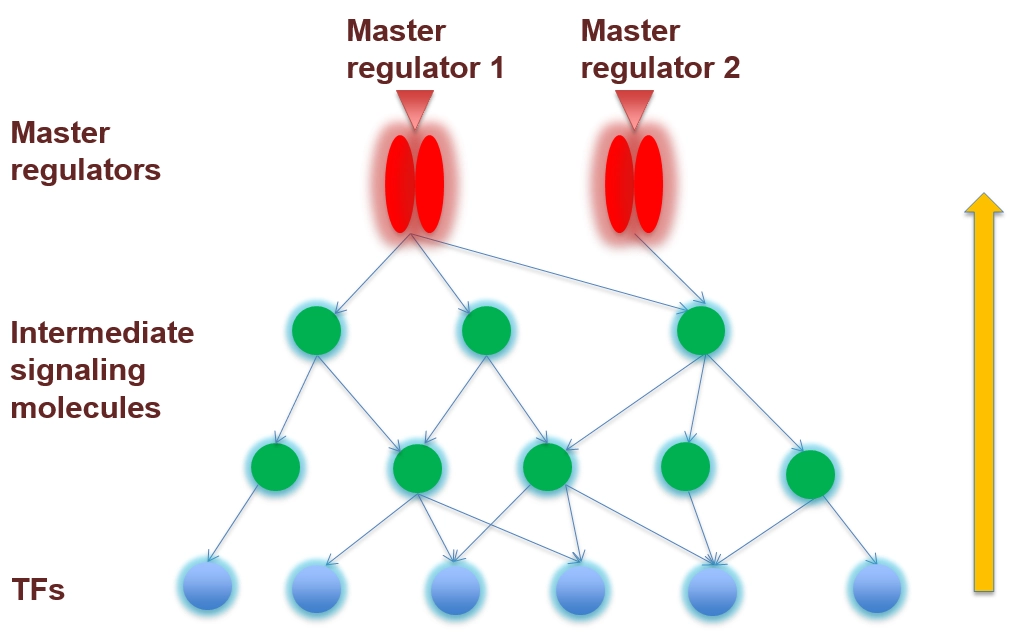
Activities of transcription factors (TFs, blue circles) are regulated by upstream signaling cascades (components shown as green circles). These converge in certain nodes, representing molecules that are potential master regulators of the process under study.