PASS
Prediction of Activity Spectra for Substances
Compute biological activity for chemical compounds using the SAR (Structure–Activity Relationship) approach
Introduction
PASS (Prediction of Activity Spectra for Substances) predicts biological activity profiles of drug-like compounds using a SAR (structure–activity relationship) approach. Based on the compound’s structural formula, PASS provides a list of probable biological activities along with two probabilities:
By default, activities with Pa > Pi are considered as probable “Actives”, but users can adjust the threshold depending on specific tasks.
PASS is widely used in medicinal chemistry and pharmaceutical research, with over 1,200 scientific publications referencing its methodology and applications.

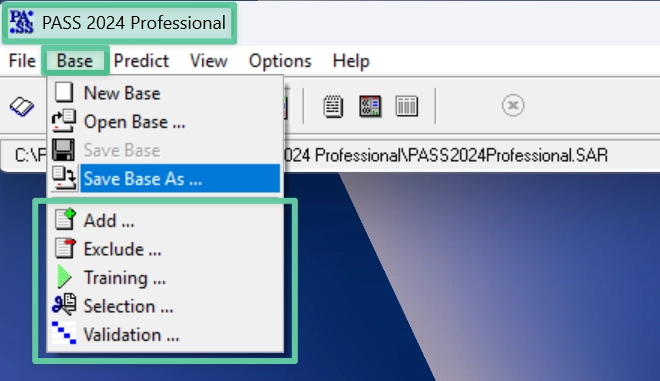
Here we show how to operate the basic interface of PASS.
PASS 2024: Current Statistics
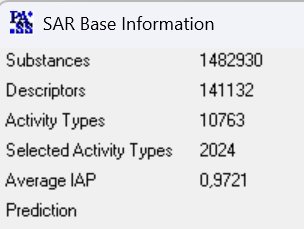
PASS-2024 SAR base is based on the information about structure-activity relationships of 1,482,930 drugs, drug-candidates, pharmaceutical agents and chemical probes as well as compounds for which known specific toxicity information.
The total number of pairwise structure-activity records is 6,019,343.
The total number of predictable activities is and 9,274 with an average accuracy over 0.934.
The recommended types of biological activities, 2,024, are predicted with an average accuracy above 0.972.
PASS 2024 Editions
PASS-2024-Standard
Predicts 2,024 recommended biological activity types (avg. accuracy > 0.972)
Total 9,274 activity types can be selected via the “Selection” procedure
PASS-2024-Refined
Includes a ready-made SAR base optimized for 2,024 recommended activities (avg. accuracy > 0.972)
PASS-2024-Professional
Includes the same SAR base as PASS-2024-Standard
Also allows training and validation of a custom SAR base using proprietary data
Your trained SAR base remains local and exclusive
Key features
Comprehensive biological activity prediction
Several thousand biological activities are predicted simultaneously, including:
Flexible selection from thousands of activity types
The number of predictable activity types is 8,565, and 1,957 activity types are in the recommended activity list. The average invariant accuracy of prediction (IAP) exceeded 0.93 for all 8,565 predictable activities, and is over 0.97 for the recommended activities. Depending on the particular purpose, the user may include into the predictable activity list any of the 8,565 activity types using the “Selection” procedure.
Focused activity selection for faster screening
Users can select pharmacotherapeutic fields or specific activity groups to focus predictions and reduce computation time.
Prediction based on structural formula only
PASS works with 2D molecular structures, making it suitable for use even with virtual (not yet synthesized) compounds.
Robust algorithm performance on incomplete datasets
The prediction engine performs reliably even when the training data is incomplete, which is common in biological research.
Prediction of adverse or toxic effects
PASS supports early-stage safety profiling by flagging potentially toxic or harmful activities.
Training of proprietary SAR bases
PASS-Professional supports training a unique SAR base from user-supplied data. This enables fully private in-house predictions.
High-throughput virtual screening
PASS is optimized for fast, large-scale predictions on libraries with millions of compounds.
New “Drug-Likeness” term
A new predictable term, “Drug-Likeness”, was added in 2024.
It was trained on 4,007 compounds with an IAP of 0.852
(Sukhachev V.S. et al., Pharmaceutical Chemistry Journal, 2024)
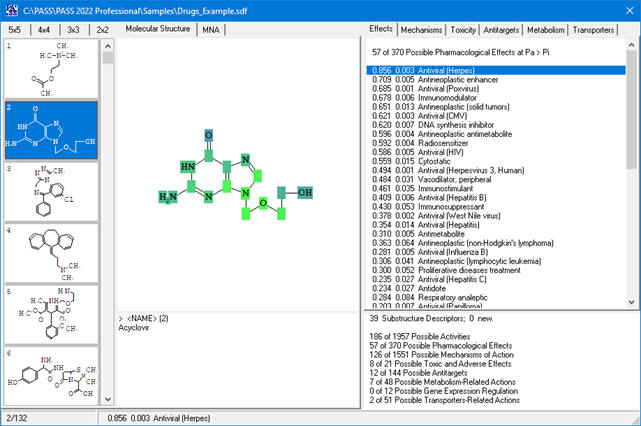
Roles of individual atoms
Based on the PASS 2024 prediction results, users can evaluate the contribution of each of the atoms of the structure to the estimated biological activity.
Once the desired biological activity in the predicted activity spectra is selected, each of the atoms in the structure is colored according to its effect
(see the detailed color-coding scheme in the ‘Roles of individual atoms’ section below)
How PASS works
PASS (Prediction of Activity Spectra for Substances) uses the structural formula of a drug-like substance as an input and computes its estimated biological activity profile (or spectrum) as an output.
The predicted biological activity list includes the names of the probable activities with two probabilities:
By default, all activities with Pa > Pi are considered as probable; however, depending on the particular tasks, the user may choose any other cutoff for selecting the probable “Actives”.
Biological activity
Biological activity is the result of a chemical compound’s interaction with biological objects.
It depends on:
In PASS, biological activities are described qualitatively as either “active” or “inactive”.
Biological activity spectrum
The Biological Activity Spectrum of a chemical compound is the set of different types of biological activity that reflect the results of the compound’s interaction with various biological entities.
It represents the “intrinsic” property of a compound depending only on its chemical structure.
Though this may be a generalization, it provides the possibility for combining information from many different sources in the same training set, which is necessary because no one particular publication comprehensively covers all the various facets of the biological action of a compound.
Chemical structure representation
The 2D structural formulae of organic compounds were chosen as the basis for the description of chemical structures, because this is the only information available at the early stage of research (compounds may only be designed but not synthesized yet).
The structure descriptors we use, which we call Multilevel Neighborhoods of Atoms (MNA), were specifically designed for chemical structure representation for SAR analysis realized in PASS and similar approaches (Filimonov D. et al., 1999).
Extended Connectivity Fingerprints (ECFP), which were developed later (Glen R.C. et al., 2006), are based on the same idea as MNA descriptors:
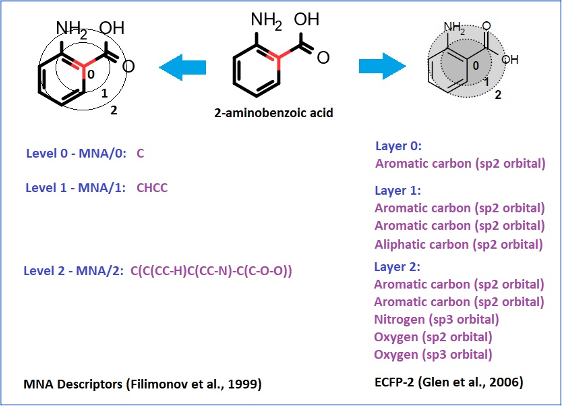
MNA descriptors, unlike ECFP, preserve the connectivity between atoms of different layers in the form of a nested bracket structure.
They are based on the molecular structure representation, which includes all hydrogen atoms according to the valences and charges of atoms and does not specify bond types.
Therefore, they inherently include the information about the type of hybridization of atomic orbitals.
Equivalent structures
The chemical compounds are considered equivalent in PASS if their molecular structures have the same MNA descriptors set.
Since MNA descriptors do not represent the stereochemical peculiarities of a molecule, structures that only differ by stereochemistry are formally considered equivalent.
SAR Base
The SAR Base contains:
Unfortunately, it is currently impossible to collect sufficiently large numbers of active compounds for all biological activities from available sources.
That is why some activity types are represented in the general PASS training set by more than 300,000 drug-like compounds, while others are represented by only a few.
The SAR Base supplied with PASS consists of substances with at least one experimentally confirmed biological activity.
PASS 2024 SAR Base is based on the information about structure–activity relationships obtained with an in-house training set of more than 1,482,930 compounds with 10,763 known biological activities.
This training set is continuously curated and expanded.
SAR Base can also be replaced by an exclusive knowledgebase, which can be created using proprietary in-house data.
SAR Base, together with the user-defined constraints of biological activities of interest and relevant parameters, provides PASS the starting point for the computational prediction.
A more detailed description of the PASS method is available in several publications, including:
Filimonov et al., 2014. (PDF file may be obtained on request).
Technical Notes
In PASS 2024, the MNA descriptors (for prediction of activity spectra or for adding substances to SAR Base) are generated if the structure corresponds to the following criteria:
All other limitations on the structural formulae implemented in the previous PASS versions
(e.g. only one uncharged component, minimum three carbon atoms in the structure, MW < 1,250)
are not applied anymore.
If the structure does not correspond to these criteria or the input data contains any other errors,
a message about the first critical error will be received.
Roles of individual atoms
Based on the PASS 2024 prediction results, the researchers can evaluate the contribution of each of the atoms to the estimated biological activity.
As soon as the desired biological activity in the predicted activity spectra is selected, each of the atoms in the structure is colored according to the following scheme:
|
Color |
Values |
Interpretation |
|
Light Green |
Pa = 1, Pi = 0 |
Atom promotes activity |
|
Light Red |
Pa = 0, Pi = 1 |
Atom promotes inactivity |
|
Light Blue |
Pa = 0, Pi = 0 |
Atom does not generate any signal |
|
Grey |
Pa = 0.33, Pi = 0.33 |
Atom equivocal for weak signal |
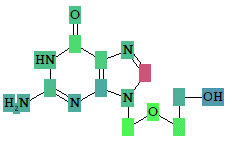
Activity prediction for a chemical substance by PASS
The slide show demonstrating the look-and-feel of PASS can be found below. A particularly useful tool to analyze and utilize PASS results further is PharmaExpert.
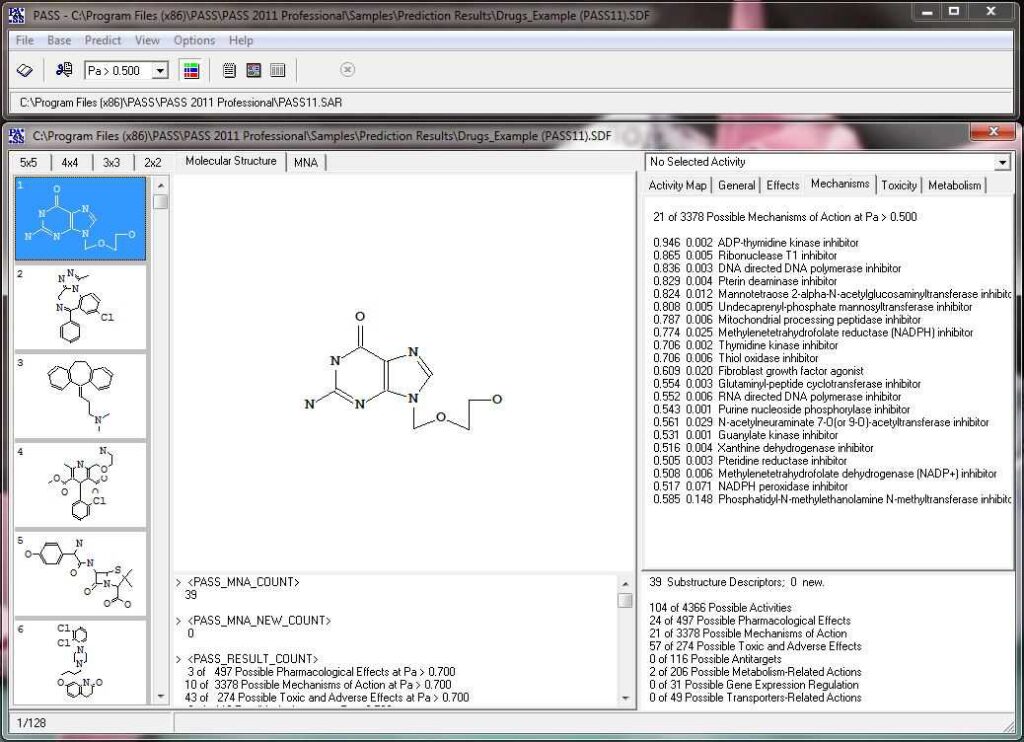
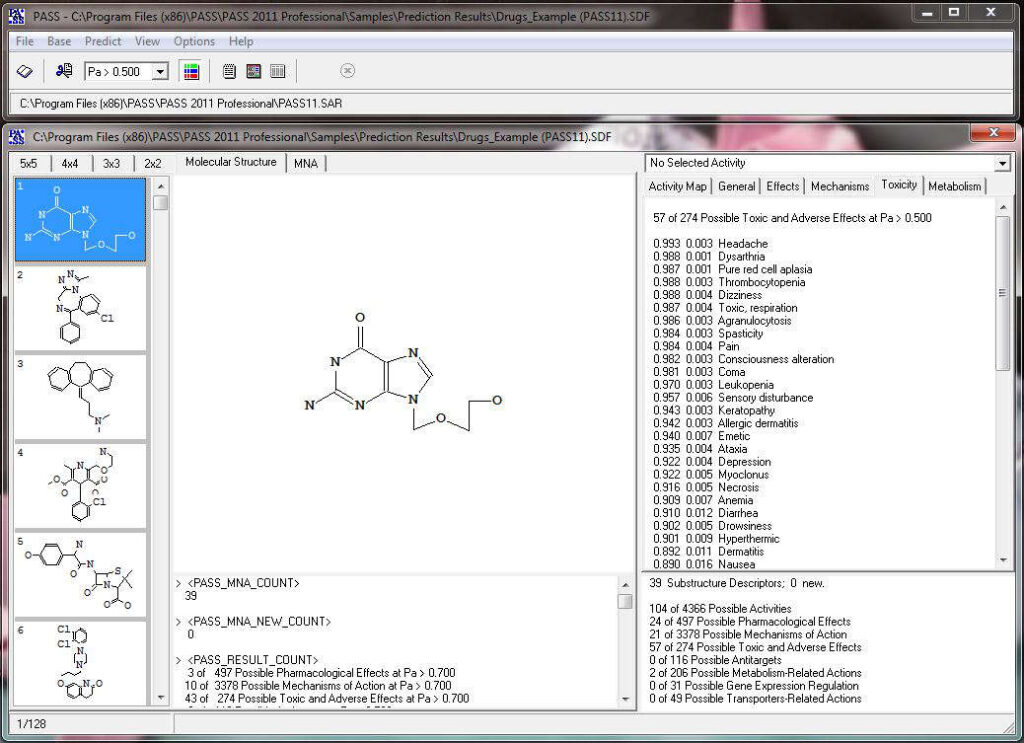
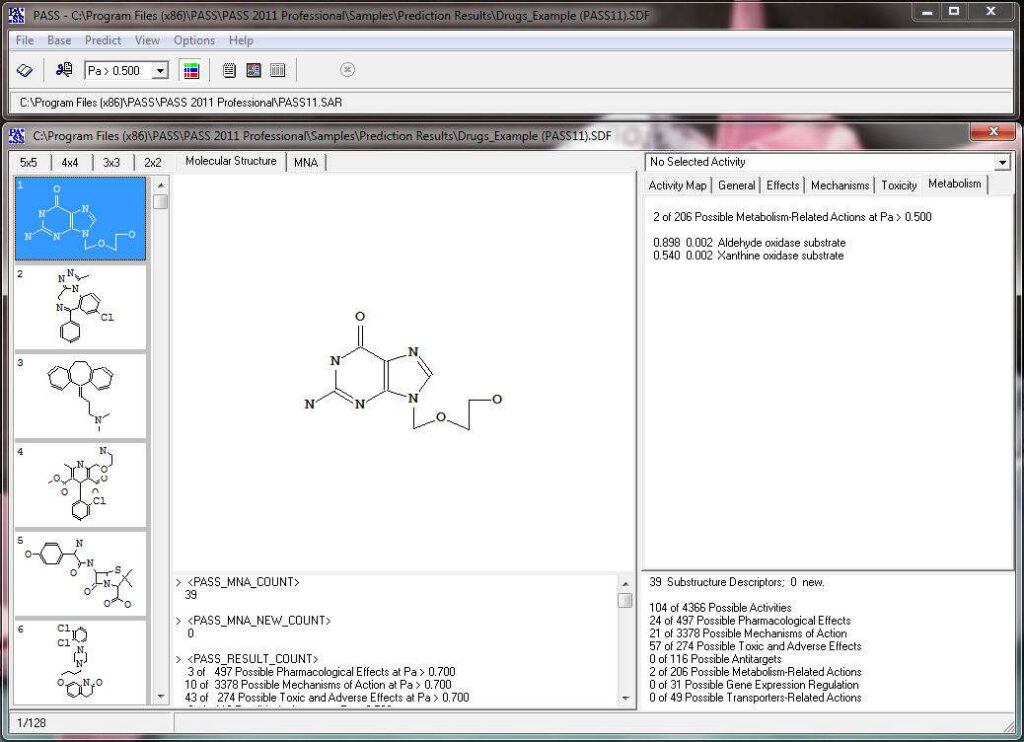

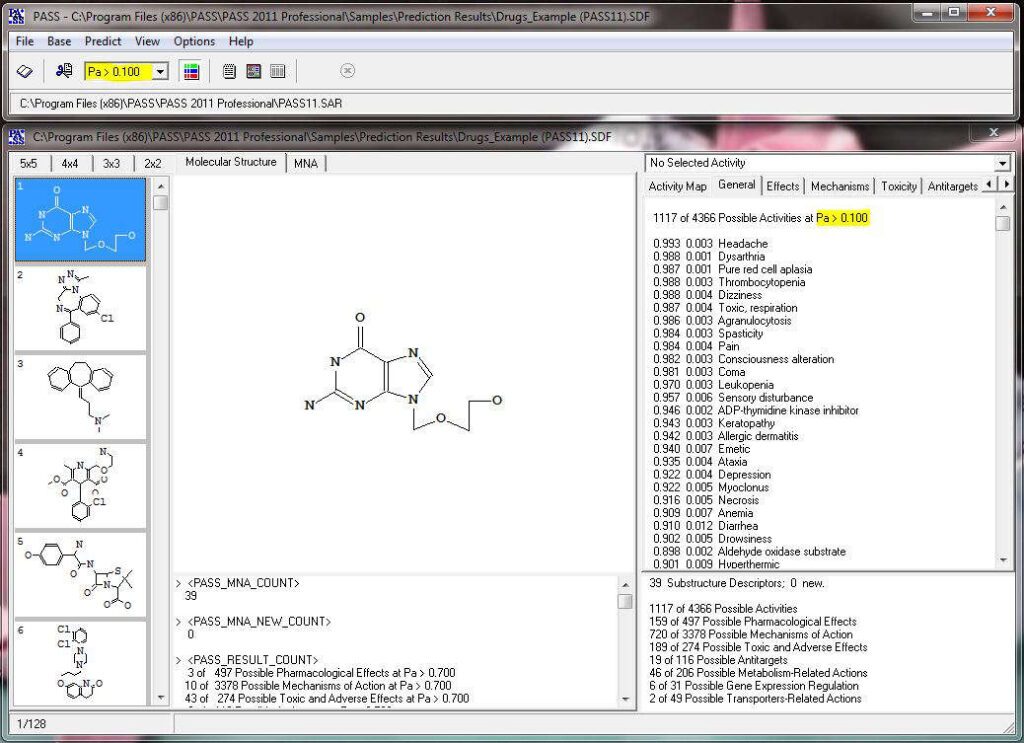
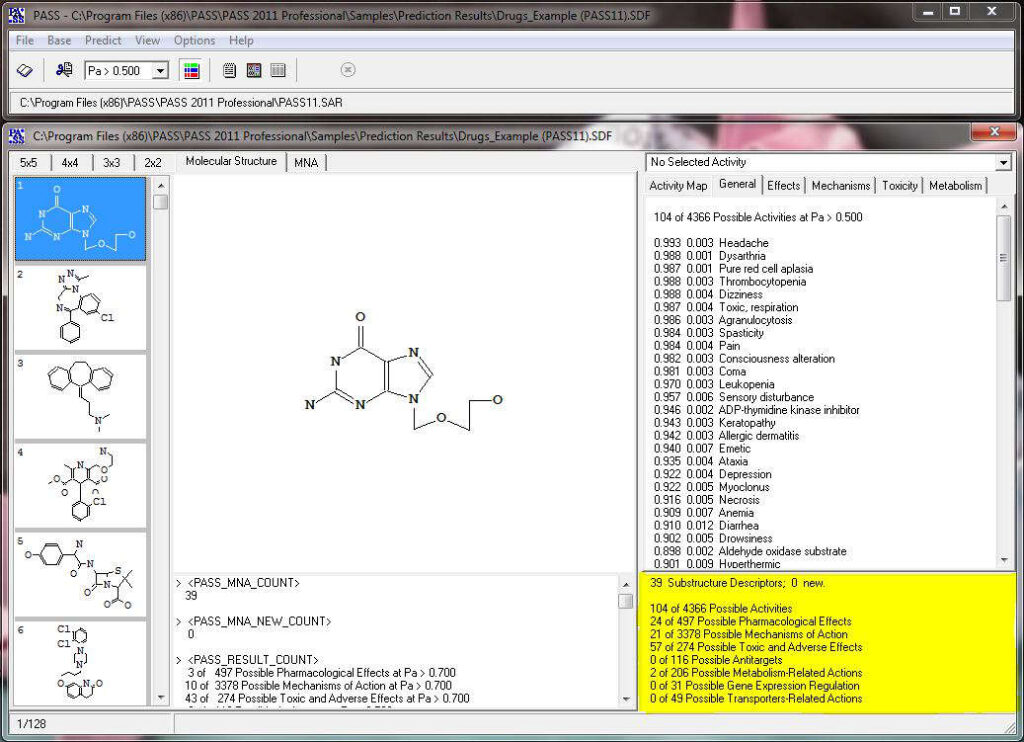
Benefits of PASS + PharmaExpert
How to cite
Lagunin A, Stepanchikova A, Filimonov D, Poroikov V.
PASS: prediction of activity spectra for biologically active substances.
Bioinformatics. 2000 Aug;16(8):747–748.
https://doi.org/10.1093/bioinformatics/16.8.747